Search Results for author:
Found 27 papers, 8 papers with code
A prediction rigidity formalism for low-cost uncertainties in trained neural networks
Regression methods are fundamental for scientific and technological applications.
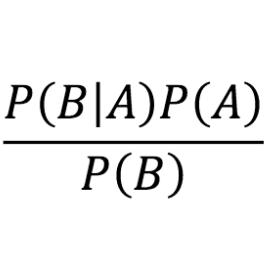

Electronic excited states from physically-constrained machine learning
Data-driven techniques are increasingly used to replace electronic-structure calculations of matter.
Physics-inspired Equivariant Descriptors of Non-bonded Interactions
One essential ingredient in many machine learning (ML) based methods for atomistic modeling of materials and molecules is the use of locality.
Smooth, exact rotational symmetrization for deep learning on point clouds
Point clouds are versatile representations of 3D objects and have found widespread application in science and engineering.
Wigner kernels: body-ordered equivariant machine learning without a basis
Machine-learning models based on a point-cloud representation of a physical object are ubiquitous in scientific applications and particularly well-suited to the atomic-scale description of molecules and materials.
 Ranked #2 on
Formation Energy
on QM9
Ranked #2 on
Formation Energy
on QM9
Completeness of Atomic Structure Representations
In this paper, we address the challenge of obtaining a comprehensive and symmetric representation of point particle groups, such as atoms in a molecule, which is crucial in physics and theoretical chemistry.
Fast evaluation of spherical harmonics with sphericart
Spherical harmonics provide a smooth, orthogonal, and symmetry-adapted basis to expand functions on a sphere, and they are used routinely in physical and theoretical chemistry as well as in different fields of science and technology, from geology and atmospheric sciences to signal processing and computer graphics.
A data-driven interpretation of the stability of molecular crystals
Due to the subtle balance of intermolecular interactions that govern structure-property relations, predicting the stability of crystal structures formed from molecular building blocks is a highly non-trivial scientific problem.
A smooth basis for atomistic machine learning
Machine learning frameworks based on correlations of interatomic positions begin with a discretized description of the density of other atoms in the neighbourhood of each atom in the system.
Electronic-structure properties from atom-centered predictions of the electron density
The electron density of a molecule or material has recently received major attention as a target quantity of machine-learning models.
Predicting hot-electron free energies from ground-state data
Machine-learning potentials are usually trained on the ground-state, Born-Oppenheimer energy surface, which depends exclusively on the atomic positions and not on the simulation temperature.
Unified theory of atom-centered representations and message-passing machine-learning schemes
Data-driven schemes that associate molecular and crystal structures with their microscopic properties share the need for a concise, effective description of the arrangement of their atomic constituents.
Incompleteness of graph neural networks for points clouds in three dimensions
We construct pairs of distinct point clouds whose associated graphs are, for any cutoff radius, equivalent based on a first-order Weisfeiler-Lehman test.
Equivariant representations for molecular Hamiltonians and N-center atomic-scale properties
Symmetry considerations are at the core of the major frameworks used to provide an effective mathematical representation of atomic configurations that is then used in machine-learning models to predict the properties associated with each structure.
Optimal radial basis for density-based atomic representations
For each training dataset and number of basis functions, one can determine a unique basis that is optimal in this sense, and can be computed at no additional cost with respect to the primitive basis by approximating it with splines.
Physics-inspired structural representations for molecules and materials
The first step in the construction of a regression model or a data-driven analysis, aiming to predict or elucidate the relationship between the atomic scale structure of matter and its properties, involves transforming the Cartesian coordinates of the atoms into a suitable representation.
Chemical Physics
Improving Sample and Feature Selection with Principal Covariates Regression
Selecting the most relevant features and samples out of a large set of candidates is a task that occurs very often in the context of automated data analysis, where it can be used to improve the computational performance, and also often the transferability, of a model.
Machine learning at the atomic-scale
Statistical learning algorithms are finding more and more applications in science and technology.
Chemical Physics Computational Physics
Uncertainty estimation for molecular dynamics and sampling
Given the interpolative nature of these models, the reliability of predictions depends on the position in phase space, and it is crucial to obtain an estimate of the error that derives from the finite number of reference structures included during the training of the model.
The role of feature space in atomistic learning
In this work we introduce a framework to compare different sets of descriptors, and different ways of transforming them by means of metrics and kernels, in terms of the structure of the feature space that they induce.
Multi-scale approach for the prediction of atomic scale properties
Electronic nearsightedness is one of the fundamental principles governing the behavior of condensed matter and supporting its description in terms of local entities such as chemical bonds.
Recursive evaluation and iterative contraction of $N$-body equivariant features
While it has become clear that low-order density correlations do not provide a complete representation of an atomic environment, the exponential increase in the number of possible $N$-body invariants makes it difficult to design a concise and effective representation.
Chemical Physics
Learning the electronic density of states in condensed matter
The electronic density of states (DOS) quantifies the distribution of the energy levels that can be occupied by electrons in a quasiparticle picture, and is central to modern electronic structure theory.
Predicting molecular dipole moments by combining atomic partial charges and atomic dipoles
In this work, we choose to represent this quantity with a physically inspired ML model that captures two distinct physical effects: local atomic polarization is captured within the symmetry-adapted Gaussian process regression (SA-GPR) framework, which assigns a (vector) dipole moment to each atom, while movement of charge across the entire molecule is captured by assigning a partial (scalar) charge to each atom.
Structure-Property Maps with Kernel Principal Covariates Regression
Data analyses based on linear methods constitute the simplest, most robust, and transparent approaches to the automatic processing of large amounts of data for building supervised or unsupervised machine learning models.
Ab initio thermodynamics of liquid and solid water
Thermodynamic properties of liquid water as well as hexagonal (Ih) and cubic (Ic) ice are predicted based on density functional theory at the hybrid-functional level, rigorously taking into account quantum nuclear motion, anharmonic fluctuations and proton disorder.
Materials Science Statistical Mechanics Chemical Physics
Comparing molecules and solids across structural and alchemical space
For instance, a structural similarity metric is crucial for classifying structures, searching chemical space for better compounds and materials, and driving the next generation of machine-learning techniques for predicting the stability and properties of molecules and materials.
Materials Science Chemical Physics
