Molecular Docking
21 papers with code • 0 benchmarks • 0 datasets
Predicting the binding structure of a small molecule ligand to a protein, which is critical to drug design.
Description from: DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Benchmarks
These leaderboards are used to track progress in Molecular Docking
Libraries
Use these libraries to find Molecular Docking models and implementations
Most implemented papers
Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity

The atomic convolutional neural network is trained to predict the experimentally determined binding affinity of a protein-ligand complex by direct calculation of the energy associated with the complex, protein, and ligand given the crystal structure of the binding pose.
SHREC 2022: Protein-ligand binding site recognition
This paper presents the methods that have participated in the SHREC 2022 contest on protein-ligand binding site recognition.
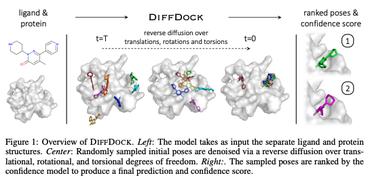
DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
gcorso/diffdock
•
 •
•
We instead frame molecular docking as a generative modeling problem and develop DiffDock, a diffusion generative model over the non-Euclidean manifold of ligand poses.
Using the Fast Fourier Transform in Binding Free Energy Calculations
According to implicit ligand theory, the standard binding free energy is an exponential average of the binding potential of mean force (BPMF), an exponential average of the interaction energy between the ligand apo ensemble and a rigid receptor.
DeepAtom: A Framework for Protein-Ligand Binding Affinity Prediction
The cornerstone of computational drug design is the calculation of binding affinity between two biological counterparts, especially a chemical compound, i. e., a ligand, and a protein.
DEEPScreen: high performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations
cansyl/DEEPScreen
•
•
Chemical Science 2020
The identification of physical interactions between drug candidate compounds and target biomolecules is an important process in drug discovery.
Assigning Confidence to Molecular Property Prediction
Introduction: Computational modeling has rapidly advanced over the last decades, especially to predict molecular properties for chemistry, material science and drug design.
DOCKSTRING: easy molecular docking yields better benchmarks for ligand design
The field of machine learning for drug discovery is witnessing an explosion of novel methods.
Direct Molecular Conformation Generation
directmolecularconfgen/dmcg
•
•
Molecular conformation generation aims to generate three-dimensional coordinates of all the atoms in a molecule and is an important task in bioinformatics and pharmacology.
DEL-Dock: Molecular Docking-Enabled Modeling of DNA-Encoded Libraries
Computational models have been deployed to learn the latent binding affinities that are correlated to the sequenced count data; however, this correlation is often obfuscated by various sources of noise introduced in its complicated data-generation process.
