Grappa -- A Machine Learned Molecular Mechanics Force Field
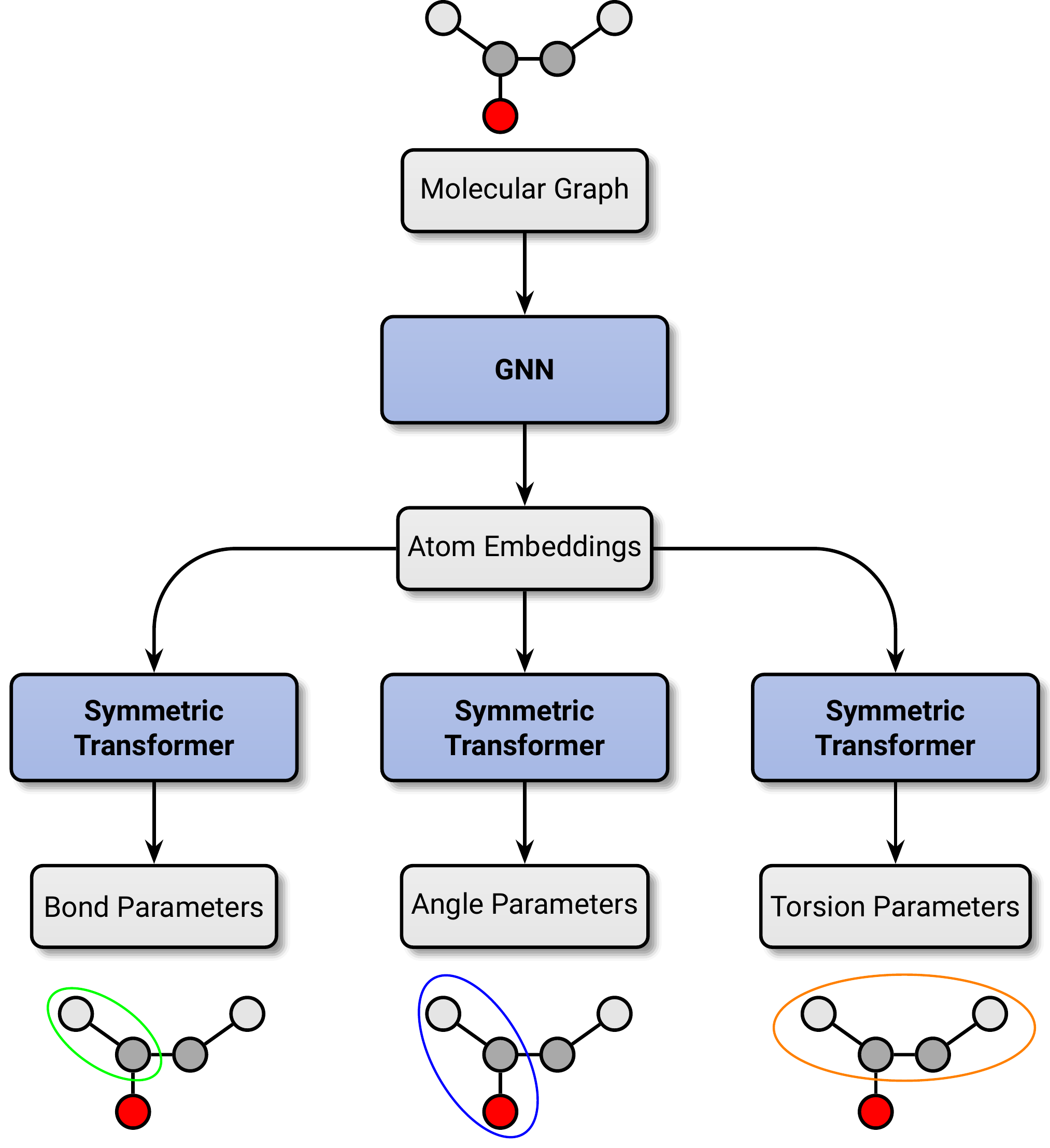
Simulating large molecular systems over long timescales requires force fields that are both accurate and efficient. In recent years, E(3) equivariant neural networks have lifted the tension between computational efficiency and accuracy of force fields, but they are still several orders of magnitude more expensive than classical molecular mechanics (MM) force fields. Here, we propose a novel machine learning architecture to predict MM parameters from the molecular graph, employing a graph attentional neural network and a transformer with symmetry-preserving positional encoding. The resulting force field, Grappa, outperforms established and other machine-learned MM force fields in terms of accuracy at the same computational efficiency and can be used in existing Molecular Dynamics (MD) engines like GROMACS and OpenMM. It predicts energies and forces of small molecules, peptides, RNA and - showcasing its extensibility to uncharted regions of chemical space - radicals at state-of-the-art MM accuracy. We demonstrate Grappa's transferability to macromolecules in MD simulations, during which large protein are kept stable and small proteins can fold. Our force field sets the stage for biomolecular simulations close to chemical accuracy, but with the same computational cost as established protein force fields.
PDF Abstract

